
Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Lissencefalie van de hersenen
Medisch expert van het artikel

Laatst beoordeeld: 12.07.2025
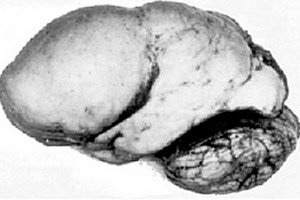
Onder de organische cerebrale pathologieën valt een aangeboren afwijking in de ontwikkeling van de hersenen als lissencefalie op, waarvan de essentie ligt in het bijna gladde oppervlak van de cortex van de hemisferen – met een onvoldoende aantal windingen en groeven. [ 1 ]
Bij volledige afwezigheid van windingen is er sprake van agyrie, en de aanwezigheid van meerdere brede, vlakke windingen wordt pachygyrie genoemd. Deze defecten hebben, net als sommige andere reductiedeformaties van de hersenen, de code Q04.3 in ICD-10.
Epidemiologie
Volgens statistieken over zeldzame ziekten zijn er 1 tot 1,2 gevallen van lissencefalie per 100 duizend pasgeborenen. [ 2 ], [ 3 ]
Volgens sommige gegevens worden bij kinderen met het Miller-Dieker-syndroom 25 tot 30% van de gevallen van klassieke lissencefalie waargenomen; puntmutaties en deleties van de LIS1- en DCX-genen worden bij bijna 85% van de patiënten aangetroffen. [ 4 ]
Uit genetische studies van 17 genen die verband houden met lissencefalie is gebleken dat 40% van de patiënten een LIS1-mutatie of -deletie heeft, en 23% een DCX-mutatie, gevolgd door TUBA1A (5%) en DYNC1H1 (3%).[ 5 ]
Oorzaken lissencefalie
Alle bekende oorzaken van de vorming van de hersenschors (cortex cerebri) vrijwel of volledig zonder windingen en groeven, die het "werkgebied" van de menselijke hersenen vergroten en de "productiviteit" van het centrale zenuwstelsel verzekeren, worden geassocieerd met verstoringen in de perinatale ontwikkeling ervan. Dat wil zeggen, lissencefalie ontwikkelt zich bij de foetus. [ 6 ]
Het onvermogen om lagen van de hersenschors van de foetus te vormen bij lissencefalie is het gevolg van abnormale migratie van de neuronen waaruit de hersenschors bestaat, of van een voortijdige beëindiging van dit proces.
Dit proces, dat essentieel is voor cerebrocorticale histogenese, vindt plaats in verschillende fasen van de 7e tot de 18e week van de zwangerschap. En gezien de verhoogde gevoeligheid voor genetische mutaties, evenals verschillende negatieve fysieke, chemische en biologische invloeden, kan elke afwijking van de norm leiden tot een onjuiste lokalisatie van neuronen met de mogelijke vorming van een verdikte laag grijze stof van de cortex zonder karakteristieke structuur. [ 7 ]
In sommige gevallen wordt lissencefalie bij kinderen in verband gebracht met het Miller-Dieker-, Walker-Warburg- of Norman-Roberts-syndroom.
Lees ook – Ontwikkelingsstoornissen van de hersenen
Risicofactoren
Naast mutaties in sommige genen behoren zuurstofgebrek (hypoxie) van de foetus tot de risicofactoren voor de geboorte van een kind met een dergelijk ernstig defect, waaronder onvoldoende bloedtoevoer naar de hersenen (hypoperfusie); een acuut cerebrovasculair accident in de vorm van een perinatale beroerte; placenta-pathologieën; virusinfecties bij de zwangere vrouw (waaronder TORCH); [ 8 ] problemen met de algemene stofwisseling en de schildklierfunctie; roken, alcoholgebruik, psychotrope en verdovende middelen; het gebruik van een aantal medicijnen; verhoogde stralingsniveaus. [ 9 ]
Pathogenese
Niet alle gevallen van lissencefalie worden veroorzaakt door chromosomale afwijkingen en genmutaties. Er zijn echter wel genen bekend die eiwitten coderen die een belangrijke rol spelen bij de correcte beweging van neuroblasten en neuronen langs de radiale gliacellen – om de hersenschors te vormen. Mutaties van deze genen leiden tot deze pathologie. [ 10 ]
In het bijzonder gaat het om sporadische mutaties (zonder erfelijkheid) van het LIS1-gen op chromosoom 17, dat het cytoplasmatische motoreiwit van microtubuli dyneïne reguleert, en van het DCX-gen op het X-chromosoom, dat codeert voor het eiwit doublecortine (lissencephalin-X). [ 11 ]. In het eerste geval definiëren specialisten klassieke lissencephalie (type I), in het tweede X-gebonden. [ 12 ]
Wanneer het FLN1-gen, dat codeert voor het fosfoproteïne filamine 1, wordt verwijderd, kan het proces van gerichte neuronale migratie helemaal niet beginnen, wat leidt tot een volledige afwezigheid van convoluties (agyrie). [ 13 ]
Er zijn mutaties geïdentificeerd in het CDK5-gen, dat codeert voor een kinase-enzym – een katalysator voor intracellulair metabolisme, dat de celcyclus in neuronen in het centrale zenuwstelsel reguleert en hun normale migratie tijdens de prenatale vorming van hersenstructuren garandeert.
Abnormale veranderingen in het RELN-gen op chromosoom 7 die defecten in de corticale gyrale cortex veroorzaken bij het Norman-Roberts-syndroom resulteren in een tekort aan het extracellulaire glycoproteïne reelin, dat nodig is om de migratie en positionering van neurale stamcellen te reguleren tijdens de ontwikkeling van de cortex cerebri.[ 14 ], [ 15 ], [ 16 ]
Het ARX-gen codeert voor het non-aristalens homeobox-eiwit, een transcriptiefactor die een belangrijke rol speelt in de voorhersenen en andere weefsels.[ 17 ] Kinderen met de ARX-mutatie hebben andere symptomen, zoals ontbrekende delen van de hersenen (agenesie van het corpus callosum), abnormale genitaliën en ernstige epilepsie.[ 18 ],[ 19 ]
Verschillende genen zijn in verband gebracht met lissencefalie. Deze genen omvatten VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A en CDK5.[ 20 ]
Cytomegalovirus (CMV) is in verband gebracht met de ontwikkeling van lissencefalie als gevolg van een verminderde bloedtoevoer naar de hersenen van de foetus. De ernst van een CMV-infectie hangt af van de zwangerschapsduur. Vroege infectie veroorzaakt vaker lissencefalie omdat neuronale migratie vroeg in de zwangerschap plaatsvindt.[ 21 ]
Bovendien omvat het ontstaansmechanisme van deze afwijking een onvolledige of latere stopzetting van de migratie van neuronen van de periventriculaire generatieve zone naar de hersenschors. In dergelijke gevallen ontwikkelt zich ofwel onvolledige lissencefalie ofwel pachygyrie, waarbij zich verschillende brede groeven en windingen vormen (maar de meeste daarvan ontbreken).
Symptomen lissencefalie
De eerste tekenen van deze pathologie (bij afwezigheid van de eerder genoemde syndromen) manifesteren zich mogelijk niet direct na de geboorte, maar pas na anderhalve tot twee maanden. Meestal worden de volgende klinische symptomen van lissencefalie waargenomen:
- spierhypotonie, vaak gecombineerd met spastische verlamming;
- convulsies en gegeneraliseerde tonisch-clonische aanvallen (in de vorm van opisthotonus);
- ernstige mentale retardatie en groeiachterstand;
- aantasting van neurologische en motorische functies.
Problemen met slikken veroorzaken moeilijkheden bij het voeden van de baby. [ 22 ]
Een ernstige neuromotorische stoornis manifesteert zich vaak als tetraplegie – verlamming van alle ledematen. Misvorming van handen, vingers of tenen is mogelijk.
Bij het Norman-Roberts-syndroom met lissencefalie type I worden craniofaciale afwijkingen waargenomen: ernstige microcefalie, lage voorhoofdshelling en uitstekende brede neusbrug, wijd uit elkaar staande ogen (hyperterlorisme), onderontwikkeling van de kaken (micrognathie). [ 23 ]
Het Miller-Dieker-syndroom kan ook worden gekenmerkt door een abnormaal klein hoofd met een breed, hoog voorhoofd en een korte neus, kuiltjes in de slapen (bitemporale kuiltjes) en laagstaande, misvormde oren.
Ernstig lissencefaliesyndroom wordt gekenmerkt door microcefalie, een afname van de grootte van de oogbollen (microftalmie), gecombineerd met netvliesdysplasie, obstructieve hydrocefalie en een afwezig of hypoplastisch corpus callosum.
Complicaties en gevolgen
Tot de complicaties van deze afwijking rekenen specialisten onder meer een verstoring van de slikfunctie (dysfagie) en gastro-oesofageale reflux; refractaire (ongecontroleerde) epilepsie; frequente infecties van de bovenste luchtwegen; longontsteking (inclusief chronische aspiratie).
Bij baby's met lissencefalie kunnen aangeboren organische hartproblemen voorkomen in de vorm van een atriumseptumdefect of een complex hartdefect met cyanose (tetralogie van Fallot). [ 24 ]
De gevolgen van postnatale groeivertraging resulteren in de meeste gevallen in de dood binnen 24 maanden na de geboorte.
Diagnostics lissencefalie
De diagnose begint met een lichamelijk onderzoek van het kind, een bestudering van de medische voorgeschiedenis van de ouders en de zwangerschap en bevalling.
Tijdens de zwangerschap kan een celvrije foetale DNA-test, een vruchtwaterpunctie of een vlokkentest nodig zijn. [ 25 ] Zie voor meer informatie – Prenatale diagnose van aangeboren aandoeningen
Instrumentele diagnostiek wordt gebruikt om hersenstructuren te visualiseren en hun functies te beoordelen:
- computertomografie van de hersenen;
- Magnetic resonance imaging (MRI) van de hersenen;
- elektro-encefalogram (EEG). [ 26 ]
Tijdens de zwangerschap kan bij een echo van de foetus na 20-21 weken lissencefalie worden vermoed, aangezien de parieto-occipitale en calcarinusgroeven ontbreken en er een afwijking is van de fissura Sylvius in de hersenen.
Differentiële diagnose
Er wordt differentiële diagnostiek met andere syndromen van aangeboren hersenafwijkingen uitgevoerd.
Er zijn meer dan twintig typen lissencefalie, waarvan de meeste in twee hoofdcategorieën vallen: klassieke lissencefalie (type 1) en cobblestone-lissencefalie (type 2). Elke categorie heeft vergelijkbare klinische manifestaties, maar verschillende genetische mutaties.[ 27 ]
Hersenonderzoek bij type I lissencefalie toont een hersenschors met vier lagen in plaats van de zes die bij normale patiënten voorkomen, terwijl bij type 2 lissencefalie de hersenschors gedesorganiseerd is en er hobbelig of nodulair uitziet als gevolg van volledige verplaatsing van de hersenschors door clusters van corticale neuronen, gescheiden door gliomesenchymweefsel. Patiënten hadden ook spier- en oogafwijkingen.
- Klassieke lissencefalie (type 1):
- LIS1: Geïsoleerde lissencefalie en Miller-Dieker-syndroom (lissencefalie geassocieerd met gezichtsdysmorfie). [ 28 ]
- LISX1: DCX-genmutatie. Vergeleken met lissencefalie veroorzaakt door LIS1-mutaties, vertoont DCX een cortex met zes lagen in plaats van vier.
- Geïsoleerde lissencefalie zonder andere bekende genetische defecten
- Keisteen lissencefalie (type 2):
- Walker-Warburg-syndroom
- Fukuyama-syndroom
- Ziekte van spieren, ogen en hersenen
- Andere typen kunnen niet in een van de twee bovenstaande groepen worden geplaatst:
- LIS2: Syndroom van Norman-Roberts, vergelijkbaar met lissencefalie type I of syndroom van Miller-Dieker, maar zonder de deletie van chromosoom 17.
- LIS3
- LISX2
Microlissencefalie: Dit is een combinatie van de afwezigheid van normale corticale plooiing en een abnormaal klein hoofd. Kinderen met normale lissencefalie hebben bij de geboorte een normaal hoofd. Kinderen met een klein hoofd bij de geboorte krijgen meestal de diagnose microlissencefalie.
Het is ook belangrijk om onderscheid te maken tussen lissencefalie en polymicrogyrie; dit zijn twee verschillende ontwikkelingsstoornissen in de hersenen.
Met wie kun je contact opnemen?
Behandeling lissencefalie
Lissencefalie is een ongeneeslijk organisch defect, dus alleen ondersteunende en symptomatische behandeling is mogelijk. [ 29 ]
Allereerst zijn dit het gebruik van anti-epileptica en anti-epileptica, en het plaatsen van een gastrostomiesonde in de maag (als het kind niet zelfstandig kan slikken). Massage is ook nuttig.
Bij ernstige hydrocefalie wordt hersenvocht afgenomen.
Het voorkomen
Deskundigen adviseren aanstaande ouders om genetisch advies in te winnen en dat zwangere vrouwen zich tijdig melden bij gynaecologen en verloskundigen. Ook moeten zij alle geplande onderzoeken ondergaan.
Prognose
Bij kinderen met lissencefalie hangt de prognose af van de ernst ervan, maar meestal overschrijdt de mentale ontwikkeling van het kind het niveau van vier tot vijf maanden niet. En alle kinderen met deze diagnose lijden aan ernstige psychomotorische stoornissen en moeilijk te behandelen epilepsie. [ 30 ]
Volgens het NINDS (het Amerikaanse National Institute of Neurological Disorders and Stroke) bedraagt de maximale levensverwachting voor mensen met lissencefalie ongeveer 10 jaar.