
Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Martin-Bell-syndroom
Medisch expert van het artikel

Laatst beoordeeld: 04.07.2025
Het Martin-Bell-syndroom werd in 1943 beschreven door artsen, naar wie het vernoemd is. De ziekte is een genetische aandoening die bestaat uit mentale retardatie. In 1969 werden veranderingen in chromosoom X (fragiliteit in de distale arm) vastgesteld die kenmerkend zijn voor deze ziekte. In 1991 ontdekten wetenschappers het gen dat verantwoordelijk is voor het ontstaan van deze ziekte. Deze ziekte wordt ook wel het "fragiele X-syndroom" genoemd. Zowel jongens als meisjes zijn vatbaar voor de ziekte, maar jongens worden er vaker (3 keer) door getroffen.
Epidemiologie
Het Martin-Bell-syndroom is een vrij veel voorkomende ziekte: 0,3-1,0 op de 1000 mannen en 0,2-0,6 op de 1000 vrouwen lijden aan deze ziekte. Bovendien worden kinderen met het Martin-Bell-syndroom op alle continenten even vaak geboren. Uiteraard hebben nationaliteit, huidskleur, oogvorm, leefomstandigheden en het welzijn van de persoon geen invloed op het voorkomen van de ziekte. De frequentie ervan is slechts vergelijkbaar met die van het Downsyndroom (1 op de 600-800 pasgeborenen). Een vijfde van de mannelijke dragers van het afwijkende gen is gezond, heeft geen klinische of genetische afwijkingen, de rest vertoont tekenen van mentale retardatie, variërend van milde tot ernstige vormen. Van de vrouwelijke dragers is iets meer dan een derde ziek.
Het fragiele X-syndroom treft ongeveer 1 op de 2500-4000 mannen en 1 op de 7000-8000 vrouwen. De dragerschapsprevalentie onder vrouwen wordt geschat op 1 op de 130-250; onder mannen op 1 op de 250-800.
Oorzaken Martin-Bell-syndroom
Het Martin-Bell-syndroom ontwikkelt zich door de volledige of gedeeltelijke stopzetting van de aanmaak van een specifiek eiwit door het lichaam. Dit gebeurt door een gebrek aan respons van het FMR1-gen, gelokaliseerd in het X-chromosoom. De mutatie treedt op als gevolg van de herstructurering van het gen vanuit onstabiele structurele varianten van de genstatussen (allelen), en niet vanaf het allereerste begin. De ziekte wordt alleen via de mannelijke lijn overgedragen en de man hoeft niet per se ziek te zijn. Mannelijke dragers geven het gen ongewijzigd door aan hun dochters, waardoor hun mentale retardatie niet zichtbaar is. Bij verdere overdracht van het gen van de moeder op haar kinderen muteert het gen en verschijnen alle kenmerkende symptomen van deze ziekte.
Pathogenese
De pathogenese van het Martin-Bell-syndroom is gebaseerd op mutaties in het genenapparaat, die leiden tot een blokkade van de productie van FMR-eiwit, een eiwit dat essentieel is voor het lichaam, met name in neuronen, en dat aanwezig is in verschillende weefsels. Onderzoek toont aan dat FMR-eiwitten direct betrokken zijn bij de regulatieprocessen van translaties in hersenweefsel. De afwezigheid van dit eiwit of de beperkte aanmaak ervan door het lichaam leidt tot mentale retardatie.
In de pathogenese van de ziekte wordt genhypermethylering beschouwd als een sleutelstoornis. Het is echter nog niet mogelijk gebleken het ontwikkelingsmechanisme van deze stoornis definitief te identificeren.
Tegelijkertijd werd ook locusheterogeniteit van de pathologie ontdekt, die geassocieerd is met polyallellisme en polylocus. De aanwezigheid van allelische varianten van de ziekteontwikkeling werd vastgesteld, die worden veroorzaakt door het bestaan van puntmutaties en de vernietiging van het FMRL-type gen.
Patiënten hebben ook twee fragiele drielingen die gevoelig zijn voor foliumzuur, gelegen op 300 kb, en 1,5-2 miljoen bp van de fragiele drieling die het FMR1-gen bevat. Het mechanisme van mutaties in de FRAXE- en FRAXF-genen (die geïdentificeerd zijn in de bovengenoemde fragiele drielingen) is gerelateerd aan het mechanisme van aandoeningen bij het Martin Bell-syndroom. Dit mechanisme wordt veroorzaakt door de verspreiding van GCC- en CGG-herhalingen, die methylering van de zogenaamde CpG-eilanden veroorzaken. Naast de klassieke vorm van de pathologie zijn er ook twee zeldzame typen die verschillen door de uitbreiding van trinucleotide-herhalingen (in de meiose bij mannen en vrouwen).
Er werd vastgesteld dat bij de klassieke vorm van het syndroom de patiënt een speciaal nucleocytoplasmatisch eiwit van het type FMR1 mist, dat de functie vervult van het binden van verschillende mRNA's. Bovendien bevordert dit eiwit de vorming van een complex dat translatieprocessen in ribosomen ondersteunt.
Symptomen Martin-Bell-syndroom
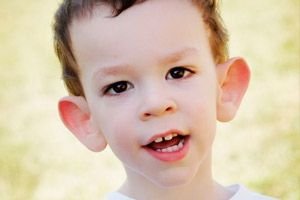
Hoe herken je de ziekte bij kinderen? Wat zijn de eerste tekenen? In de eerste maanden van het leven van een kind is het onmogelijk om het Martin-Bell-symptoom te herkennen, behalve dat er soms een afname van de spierspanning wordt waargenomen. Na een jaar is het klinische beeld van de ziekte duidelijker: het kind begint laat te lopen en te praten, soms is het spreken volledig afwezig. Het kind is hyperactief, zwaait willekeurig met zijn armen, is bang voor drukte en lawaai, is koppig, heeft heftige woede-uitbarstingen, emotionele instabiliteit, epileptische aanvallen en maakt geen oogcontact. Bij patiënten met het Martin-Bell-syndroom wordt de ziekte ook verraden door hun uiterlijk: oren zijn afstaand en groot, het voorhoofd is zwaar, het gezicht is langwerpig, de kin is vooruitgestoken, scheelzien, brede handen en voeten. Ze worden ook gekenmerkt door endocriene aandoeningen: vaak een hoog gewicht, obesitas, grote testikels bij mannen, vroege puberteit.

Bij patiënten met het Martin-Bell-syndroom varieert het intelligentieniveau sterk: van lichte tot ernstige verstandelijke beperkingen. Als een normaal persoon gemiddeld een intelligentiequotiënt (IQ) van 100 heeft en een genie 130, dan hebben mensen die vatbaar zijn voor de ziekte een intelligentiequotiënt van 35 tot 70.
Alle klinische symptomen van de pathologie kunnen worden gekarakteriseerd door een drietal hoofdverschijnselen:
- oligofrenie (IQ is 35-50);
- dysmorfofobie (afstaande oren en prognathie worden waargenomen);
- macroorchidie, dat optreedt na het begin van de puberteit.
Ongeveer 80% van de patiënten heeft ook een bicuspide klepprolaps.
De volledige vorm van het syndroom manifesteert zich echter slechts bij 60% van alle patiënten. Bij 10% wordt alleen een verstandelijke beperking vastgesteld en bij de rest ontwikkelt de ziekte zich met een andere combinatie van symptomen.
Tot de eerste tekenen van de ziekte, die zich al op jonge leeftijd openbaren, behoren:
- het zieke kind vertoont een aanzienlijke mentale retardatie in vergelijking met de ontwikkeling van de andere leeftijdsgenoten;
- aandachts- en concentratiestoornissen;
- sterke koppigheid;
- kinderen beginnen pas laat te lopen en te praten;
- er worden hyperactiviteits- en spraakontwikkelingsstoornissen waargenomen;
- zeer sterke en oncontroleerbare woedeaanvallen;
- er kan mutisme ontstaan - dit is een volledig gebrek aan spraak bij een kind;
- de baby ervaart sociale angst en kan in paniek raken bij harde geluiden of andere harde geluiden;
- het kind zwaait oncontroleerbaar en chaotisch met zijn armen;
- er wordt verlegenheid waargenomen, het kind is bang om op drukke plaatsen te zijn;
- het ontstaan van verschillende obsessieve ideeën, onstabiele emotionele toestand;
- Het kan zijn dat de baby terughoudend is om oogcontact te maken met mensen.
Bij volwassenen worden de volgende symptomen van de pathologie waargenomen:
- specifiek uiterlijk: een langwerpig gezicht met een zwaar voorhoofd, grote, uitstekende oren, een sterk uitstekende kin;
- platvoeten, middenoorontsteking en scheelzien;
- de puberteit vindt vrij vroeg plaats;
- er kan obesitas ontstaan;
- Bij het Martin-Bell-syndroom worden vrij vaak hartafwijkingen waargenomen;
- bij mannen wordt vergroting van de testikels waargenomen;
- de gewrichten worden zeer beweeglijk;
- gewicht en lengte nemen sterk toe.
Diagnostics Martin-Bell-syndroom
Om het Martin Bell-syndroom te diagnosticeren, moet u contact opnemen met een gekwalificeerde geneticus. De diagnose wordt gesteld na specifieke genetische tests waarmee het defecte chromosoom kan worden geïdentificeerd.
Testen
In een vroeg stadium van de ziekte wordt een cytogenetische methode gebruikt, waarbij een fragment van celmateriaal van de patiënt wordt afgenomen, waaraan foliumzuur wordt toegevoegd om veranderingen in de chromosomen te veroorzaken. Na verloop van tijd wordt een gebied van het chromosoom geïdentificeerd waar een duidelijke verdunning optreedt - dit is een teken van de aanwezigheid van het fragiele X-syndroom.
Deze test is echter niet geschikt voor diagnose in een later stadium van de ziekte, omdat de nauwkeurigheid ervan afneemt door het wijdverbreide gebruik van multivitaminen die foliumzuur bevatten.
Geïntegreerde diagnostiek voor het Martin-Bell-syndroom is een moleculair genetisch onderzoek, waarbij het aantal zogenoemde trinucleotide-herhalingen in het gen wordt bepaald.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentele diagnostiek
Een heel specifieke methode van instrumentele diagnostiek is PCR (polymerasekettingreactie), waarmee de structuur van aminozuren in het X-chromosoom bestudeerd kan worden en daarmee vastgesteld kan worden of er sprake is van het Martin Bell-syndroom.
Er is ook een aparte, nog specifiekere methode voor pathologische diagnostiek: een combinatie van PCR en detectie met capillaire elektroforese. Deze methode is zeer nauwkeurig en detecteert chromosomale pathologie bij patiënten met primair ovariumfalen en atactisch syndroom.
De aanwezigheid van het defect kan worden vastgesteld na diagnostiek op basis van het EEG. Patiënten met deze ziekte vertonen een vergelijkbare bio-elektrische hersenactiviteit.
Welke tests zijn nodig?
Differentiële diagnose
Er zijn verschillende methoden die kunnen helpen bij het vermoeden van het syndroom:
- klinisch - 97,5% van de patiënten heeft duidelijke tekenen van mentale retardatie (matig of ernstig); 62% heeft grote, afstaande oren; 68,4% heeft een grote, uitstekende kin en voorhoofd; 68,4% van de jongens heeft vergrote testikels, 41,4% heeft spraakafwijkingen (onregelmatige spreeksnelheid, oncontroleerbaar volume, enz.);
- cytogeen - bloed wordt onderzocht op lymfocytenkweek, het aantal cellen met een fragiel X-chromosoom per 100 bestudeerde cellen wordt bepaald;
- Elektro-encefalografie: veranderingen in elektrische impulsen van de hersenen die specifiek zijn voor het Martin-Bell-syndroom worden geregistreerd.
Met wie kun je contact opnemen?
Behandeling Martin-Bell-syndroom
Bij de behandeling van volwassen patiënten worden antidepressiva met psychostimulantia gebruikt. Het medicamenteuze behandelingsproces wordt continu gevolgd door een psycholoog en psychiater. Daarnaast worden micro-injecties met geneesmiddelen zoals Cerebrolysine (of derivaten daarvan) en cytomedicijnen (zoals Solcoseryl of Lidase) uitgevoerd in privéklinieken.
Bij de ontwikkeling van het atactisch syndroom worden bloedverdunners en nootropica gebruikt. Daarnaast worden aminozuurmengsels en angioprotectieve middelen voorgeschreven. Vrouwen met primair ovariumfalen krijgen een corrigerende behandeling voorgeschreven met kruidengeneesmiddelen en oestrogenen.
Glutamine-receptorantagonisten worden ook gebruikt bij de behandeling.
Traditioneel omvat de behandeling van het Martin-Bell-syndroom het gebruik van medicijnen die de symptomen van de ziekte beïnvloeden, maar niet de oorzaak ervan. Deze therapie omvat het voorschrijven van antidepressiva, neuroleptica en psychostimulantia. Niet alle medicijnen zijn geïndiceerd voor gebruik bij kinderen, dus de lijst met medicijnen is vrij beperkt. Neuroleptica die na 3 jaar (de vroegst mogelijke leeftijd waarop ze kunnen worden voorgeschreven) kunnen worden gebruikt, zijn onder andere haloperidol in druppels en tabletten, chloorpromazine in oplossing en periciazine in druppels. De dosis haloperidol voor kinderen wordt daarom berekend op basis van het lichaamsgewicht. Voor volwassenen wordt de dosis individueel voorgeschreven. Het wordt oraal ingenomen, beginnend met 0,5-5 mg 2-3 keer per dag, vervolgens wordt de dosis geleidelijk verhoogd tot 10-15 mg. Zodra verbetering optreedt, wordt overgeschakeld naar een lagere dosis om de bereikte conditie te behouden. Bij psychomotorische agitatie wordt 5-10 mg intramusculair of intraveneus voorgeschreven, meerdere herhalingen zijn mogelijk na 30-40 minuten. De dagelijkse dosis mag niet hoger zijn dan 100 mg. Bijwerkingen zoals misselijkheid, braken, spierspasmen, verhoogde bloeddruk, hartritmestoornissen, enz. zijn mogelijk. Ouderen dienen speciale voorzorgsmaatregelen te nemen, aangezien er gevallen van plotselinge hartstilstand zijn geregistreerd en tardieve dyskinesie (onwillekeurige bewegingen) kan optreden.
Antidepressiva verhogen de activiteit van hersenstructuren, verlichten depressie en spanning en verbeteren de stemming. Deze geneesmiddelen, aanbevolen voor gebruik bij kinderen van 5 tot 8 jaar bij het Martin-Bell-syndroom, omvatten clomipromine, sertraline, fluoxetine en fluvoxamine. Fluoxetine wordt daarom 1-2 keer (bij voorkeur in de eerste helft van de dag) oraal ingenomen tijdens de maaltijd, beginnend met 20 mg per dag, indien nodig oplopend tot 80 mg. Ouderen wordt een hogere dosis dan 60 mg niet aanbevolen. De behandeling wordt bepaald door een arts, maar mag niet langer dan 5 weken duren.
Mogelijke bijwerkingen: duizeligheid, angst, oorsuizen, verlies van eetlust, tachycardie, oedeem, enz. Voorzichtigheid is geboden bij het voorschrijven aan ouderen, mensen met hart- en vaatziekten en diabetes.
Psychostimulantia zijn psychotrope medicijnen die worden gebruikt om de waarneming van externe stimuli te verbeteren. Ze verbeteren het gehoor, de reactievermogens en het gezichtsvermogen.
Diazepam wordt voorgeschreven als kalmerend middel bij neurosen, angst, epileptische aanvallen en convulsies. Het wordt oraal, intraveneus, intramusculair en rectaal (in het rectum) ingenomen. Het wordt individueel voorgeschreven, afhankelijk van de ernst van de ziekte, met de laagste doses van 5-10 mg, en 5-20 mg per dag. De behandelingsduur is 2-3 maanden. Voor kinderen wordt de dosis berekend op basis van het lichaamsgewicht en de individuele kenmerken. Bijwerkingen zijn onder andere lethargie, apathie, slaperigheid, misselijkheid en constipatie. Het is gevaarlijk om het te combineren met alcohol; verslaving aan het middel is mogelijk.
Bij de behandeling van het Martin-Bell-syndroom zijn gevallen van verbetering van de toestand gemeld, met de introductie van geneesmiddelen gemaakt van dierlijk materiaal (hersenen): cerebrolysaat, cerebrolysine, cerebrolysaat-M. De belangrijkste bestanddelen van deze geneesmiddelen zijn peptiden die de eiwitproductie in neuronen bevorderen en zo het ontbrekende eiwit aanvullen. Cerebrolysine wordt toegediend in een injectie van 5-10 ml, de behandeling bestaat uit 20-30 injecties. Het geneesmiddel wordt voorgeschreven aan kinderen vanaf één jaar, dagelijks intramusculair toegediend in een dosis van 1-2 ml gedurende een maand. Herhaalde toedieningssessies zijn mogelijk. Bijwerkingen in de vorm van koorts zijn gecontra-indiceerd voor zwangere vrouwen.
Er zijn pogingen gedaan om de ziekte met foliumzuur te behandelen, maar alleen het gedrag verbeterde (de mate van agressie en hyperactiviteit nam af, de spraak verbeterde) en er veranderde niets op intellectueel vlak. Om de ziekte te verbeteren, wordt foliumzuur voorgeschreven en zijn fysiotherapie, logopedie en pedagogische en sociale correctie geïndiceerd.
Lithiumpreparaten worden ook als effectief beschouwd, omdat ze de aanpassing van de patiënt aan de sociale omgeving en de cognitieve activiteit verbeteren. Bovendien reguleren ze zijn gedrag in de maatschappij.
Kruiden kunnen worden gebruikt als antidepressiva bij het Martin-Bell-syndroom. Kruiden die helpen bij het verlichten van spanning en angst en die de slaap verbeteren, zijn onder andere valeriaan, pepermunt, tijm, sint-janskruid en kamille. Infusies worden als volgt bereid: voor 1 theelepel gedroogde kruiden heeft u een glas kokend water nodig. De afkooksels trekken minstens 20 minuten en worden voornamelijk 's avonds voor het slapengaan of 's middags ingenomen. Een lepel honing is een goede aanvulling.
Fysiotherapiebehandeling
Om neurologische verschijnselen uit te sluiten, worden speciale fysiotherapeutische procedures uitgevoerd, zoals oefeningen in het zwembad, spierontspanning en acupunctuur.
Chirurgische behandeling
Een belangrijke fase in de behandeling wordt ook beschouwd als plastische chirurgie - operaties die het uiterlijk van de patiënt verbeteren. Plastische chirurgie van de ledematen en oorschelpen wordt uitgevoerd, evenals de genitaliën. Ook correctie van gynaecomastie met epispadias en andere uiterlijke afwijkingen wordt uitgevoerd.
Het voorkomen
De enige methode om de ziekte te voorkomen is prenatale screening van zwangere vrouwen. Er zijn speciale onderzoeken die de pathologie vroegtijdig kunnen opsporen, waarna het raadzaam is de zwangerschap af te breken. Als alternatief kan IVF worden toegepast, wat kan helpen om het kind een gezond X-chromosoom te laten erven.
Preventie voor de patiënt hangt ervan af of de genmutatie opnieuw is opgetreden of erfelijk is. Hiervoor wordt moleculair genetische diagnostiek uitgevoerd. Het feit dat de test het "fragiele X-chromosoom" bij familieleden niet aantoonde, spreekt voor de "versheid" van de mutatie, waardoor het risico op een kind met het Martin-Bell-syndroom zeer klein is. In families met zieke mensen helpt de test herhaling van gevallen te voorkomen.
Prognose
De prognose voor het Martin-Bell-syndroom is gunstig voor het leven, maar niet voor herstel. De levensverwachting hangt af van de ernst van de ziekte en de bijbehorende afwijkingen. De patiënt kan een normaal leven leiden. Bij ernstige vormen van het Martin-Bell-syndroom lopen patiënten het risico op levenslange invaliditeit.
Levensverwachting
Het syndroom van Martin Bell heeft geen ernstige negatieve gevolgen voor de gezondheid. De levensverwachting van de meeste mensen bij wie deze pathologie is vastgesteld, wijkt dan ook niet af van de standaardindicatoren.