
Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Aper-syndroom
Medisch expert van het artikel

Laatst beoordeeld: 04.07.2025

 [
[ Oorzaken Aper-syndroom
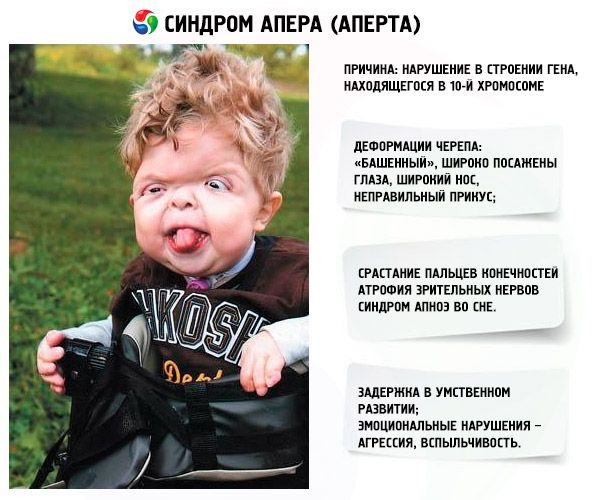
De ontwikkeling van het syndroom wordt veroorzaakt door een stoornis in de structuur van een gen dat zich op chromosoom 10 bevindt. Dit gen is verantwoordelijk voor het proces van het scheiden van de vingers en bovendien voor het tijdig sluiten van de schedelnaden.
Bovendien worden infectieziekten die tijdens de zwangerschap zijn opgelopen (zoals rodehond of tuberculose, syfilis of griep, evenals meningitis) en blootstelling van de moeder aan röntgenstraling beschouwd als oorzaken van de pathologie. Meestal wordt een dergelijk syndroom vastgesteld bij kinderen van bejaarde ouders.
Pathogenese
Het Apert-syndroom is erfelijk. Het type is autosomaal dominant (d.w.z. in een familie waarin een van de toekomstige ouders ziek is, is de kans op een kind met deze ziekte 50-100%).
Een unieke mutatie in fibroblastgroeifactorreceptor 2 (FGFR2) resulteert in een verhoogd aantal progenitorcellen die zich ontwikkelen langs de osteogene route. Dit resulteert uiteindelijk in een verhoogde subperiostale botmatrixvorming en voortijdige ossificatie van de schedelnaden tijdens de ontwikkeling van de foetus. De volgorde en snelheid van het smelten van de hechtingen bepalen de mate van misvorming en invaliditeit. Zodra het hechtingsmateriaal is genezen, wordt de groei van andere weefsels loodrecht op deze hechting beperkt en fungeren de gefuseerde botten als één enkel botplatform.
Het eerste genetische bewijs dat syndactylie bij het syndroom van Apert het gevolg is van een defect in de keratinocytgroeifactorreceptor (KGFR), was de observatie van een correlatie tussen KGFR-expressie in fibroblasten en de ernst van de syndactylie.
Amblyopie en scheelzien komen vaker voor bij patiënten met de FGFR2 Ser252Trp-mutatie, en papilatrofie komt vaker voor bij patiënten met de FGFR2 Pro253Arg-mutatie. Patiënten met FGR2 Ser252Trp-mutaties hebben een significant hogere prevalentie van visuele beperkingen in vergelijking met patiënten met de FGFR2 Pro253Arg-mutatie.
Symptomen Aper-syndroom
Sommige manifestaties van de ziekte zijn al duidelijk merkbaar bij de geboorte van de baby, omdat ze zich in de baarmoeder ontwikkelen. De belangrijkste symptomen van het syndroom zijn:
- De schedel is misvormd - hij wordt langer en krijgt het uiterlijk van een "toren". Bovendien staan de ogen wijd open en puilen ze licht uit (omdat de grootte van de oogkassen afneemt). De neus is breder geworden en er ontstaat een verkeerd gebit (de boventanden steken te ver uit).
- De vingers van de ledematen zijn volledig vergroeid (vooral de ringvinger, maar ook de middelvinger en wijsvinger) en zien eruit als een huidmembraan of een volledige vergroeiing van botten; daarnaast kunnen er extra vingers groeien;
- Verstandelijke beperking (komt niet bij iedereen voor);
- De oogzenuwen atrofiëren, waardoor de gezichtsscherpte afneemt (in sommige gevallen ontstaat er volledig gezichtsverlies);
- Verhoogde intracraniale druk, die ontstaat als gevolg van voortijdige sluiting van de schedelnaden, uit zich in de vorm van hoofdpijn en braken met misselijkheid;
- Omdat de bovenkaak onderontwikkeld blijft, ontstaan er ademhalingsproblemen;
- Slaapapneusyndroom komt vaak voor.
- Emotionele uitingen – agressie, gebrek aan zelfbeheersing, sterke opvliegendheid.
Complicaties en gevolgen
De meeste patiënten hebben in hun kindertijd een zekere mate van obstructie van de bovenste luchtwegen. De bovenste luchtwegen zijn slecht doorgankelijk vanwege de verkleinde neuskeelholte en choanae, en de onderste luchtwegen kunnen vroegtijdig overlijden veroorzaken door afwijkingen aan het tracheale kraakbeen.
Diagnostics Aper-syndroom
Om een diagnose te stellen, zijn de volgende stappen nodig:
- De arts moet de klachten van de patiënt analyseren, evenals de medische voorgeschiedenis. Het is noodzakelijk om na te gaan of er gevallen van soortgelijke pathologie in de familie voorkomen;
- Neurologisch onderzoek om de vorm van de schedel en de intellectuele ontwikkeling van de patiënt te beoordelen (speciale vragenlijsten en interview);
- Onderzoek van het fundus om de aanwezigheid van symptomen van verhoogde intracraniële druk op te sporen (zwelling van de oogzenuw, evenals vervaging van de randen ervan);
- Om de toestand van de schedel te beoordelen, wordt een röntgenfoto gemaakt;
- Computer- en magnetische resonantiebeeldvorming van het hoofd om de structuur van de hersenen en de schedel laag voor laag te onderzoeken, om de aanwezigheid van symptomen van voortijdige vergroeiing van de schedelnaden vast te stellen en bovendien hydrocefalie (als gevolg van verhoogde intracraniële druk hoopt zich overtollig hersenvocht op (dit is het hersenvocht dat het stofwisselingsproces en de voeding van de hersenen bevordert));
- Röntgenfoto van de voeten en handen om de oorzaak van de vergroeiing van de vingers te bepalen (dit is belangrijk voor de planning van een eventuele latere chirurgische ingreep);
- Mogelijk wordt een consult met een medisch geneticus en een neurochirurg voorgeschreven.
Testen
Er wordt een genetische analyse uitgevoerd van veelvoorkomende mutaties die in het gen van het type FGFR2 voorkomen.
Differentiële diagnose
Dit syndroom moet worden onderscheiden van andere genetische aandoeningen waarbij craniosynostose wordt waargenomen. Het gaat hierbij om aandoeningen zoals het syndroom van Pfeiffer, Crouzon, Saethre-Chotzen en Carpenter. Moleculair genetische testmethoden worden gebruikt om deze afwijkingen uit te sluiten.
Met wie kun je contact opnemen?
Behandeling Aper-syndroom
Een operatie wordt gezien als de enige effectieve behandeling voor het Apertsyndroom. Het helpt individuele fysieke afwijkingen te corrigeren en corrigeert ook mentale retardatie.
Tijdens deze ingreep wordt de coronale hechting gesloten om mogelijk hersenletsel te voorkomen. De meest gebruikte techniek is craniofaciale distractie, waarbij de schedel geleidelijk wordt getrokken. Orthodontische en/of orthognatische chirurgie wordt uitgevoerd om individuele gezichtsdefecten te verwijderen.
Daarnaast ondergaan patiënten een chirurgische correctie van de vingerfusie.
Referenties
Medische genetica. Nationale Gids. Bewerkt door Ginter EK, Puzyrev VP GEOTAR-Media. 2022